Documentation in general is an essential part of a quality management system (QMS). And these documents must be secure, reliable and, if necessary, have validated controls to ensure the integrity of the record throughout the retention period thus ensuring the final quality of a product or process.
And these documents, which can be electronic, manual or mixed (partly manual and partly electronic), include:
- Implemented Procedures, Communications, etc.;
- Registers in general;
- Calculations and Raw Data;
- Calibrations, Checks, Validations and Qualifications;
- Original observations;
- Results and Manual or Equipment Reports;
- Specifications, Tests, Formulas etc.;
- Master Lists.
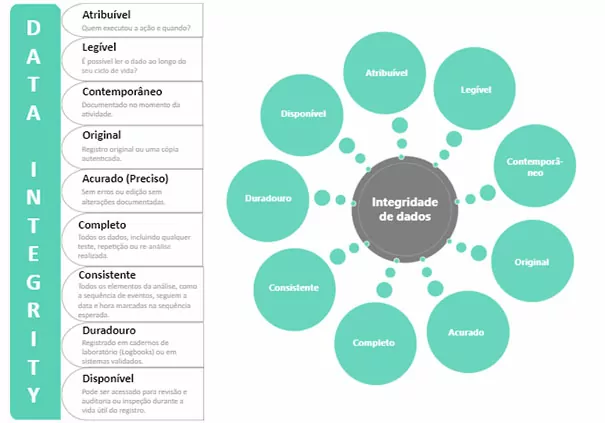
And the principle governing this part of the QMS has been established and identified by FDA (Food and Drug Administration) as ALCOA. According to this practice, a piece of data must have the following characteristics::
Attributable: the data generated or collected must be attributed to those who performed the action and when the action was performed. Examples:
- Electronic system: one must have individual logins and passwords, easily identifying who logged in, whochanged or who deleted a particular data through audit trail.
- Manual system: Records must contain dated signatures or rubrics, and all of them must be unique and recorded in a document, which can be alogbook by assigning the signature and rubric to only one person.
Legible: The collected data must be recorded legibly and permanently. Examples:
- Electronic system: records must be checked periodically for legibility[VB1] (The document in electronic medium needs to be in a format that can be opened and read by others, such as PDF.),audit trails should also be easily intelligible and unable to be modified by a user.
- Manual system: records must be made indelible and unerasable ones(records with pencils or erasable pensand use of Liquid Papers) are forbidden. In addition, errors or erasures should be corrected only with a trace, justified, signed and dated.
Contemporaneous: the data must be recorded when the action occurs. Examples:
- Electronic system: the system must automatically generate the date and time record of data entry, as well as in electronic signatures. Time changes on system clocks cannot be allowed. It is forbidden to save or forward records data.
- Manual system: records must be made at the time of activity, and retroactive records are not allowed. It is forbidden to generate photocopies of records without authorization.
Original: the data must be annotated directly in the system, onlogbookor record sheet, avoiding provisional annotations and subsequent transcription of this data. Examples:
- Electronic system: Metadata should be reviewed periodically and the system should prevent the deletion of original data.
- Manual system: the use of draft or sticky notes in the BPx area should not be allowed.
Accurate: the generated data must be error-free, complete and true, reflecting exactly the actions determined in the process. Examples:
- Electronic system: Systems must be validated and reviewed periodically. System data must be protected against unauthorized changes.
- Manual system: Deviations and results outside the specification should be investigated. Instruments should always be calibrated.It is advisable secondary checks to verify critical data accuracy and precision.
In addition to this concept, there is also ALCOA+, created by PIC/S (PharmaceuticalInspection Co-operation Scheme), whose “+” brings 4 more requirements for the qualification of data:
- Complete: Data must be complete, i.e. even invalidated tests should be included in the final report.
- Consistent: Recorded data cannot be modified throughout its lifecycle.
- Enduring: Data cannot be missed over time, for example printed data on thermal printers.
- Available: Data shall always be available throughout its life cycle, for example you must not lose data during system change.
And for a better every day view, here are some examples of data integrity failures:
- Ignore or omit certain data from a dataset;
- Intentional falsification of data (hide, replace original data, rewrite records, etc.);
- Unofficial tests;
- Deletion of failed data;
- Faking data for batch release;
- Recording samples or weights on scratch paper;
- Results outside the specification neither investigated nor unreported;
- No restrictions or protection of electronic data, access without individual passwords;
- Product released but not registered;
- Pre- or post-dated entries;
- Retroactive records;
- Use of flash drives.
And how can we prevent problems with data integrity?
- Keeping competent and trained technical personnel;
- Effective training for new employees,using the companyculture approach, correcting mindset and adjusting newly hired from companies with different cultures;;
- continued education for a good familiarization to procedures and policies;
- Awareness and correct interpretation of GMP regulations and requirements.
At all phases of a document process or lifecycle, involving data (such as: creating, writing, checking, processing, reviewing, analyzing, reporting, transferring, storing, querying, or monitoring) until it is no longer useful and becomes obsolete and ultimately deleted, data integrity must be present, either in electronic or manual format.
In addition, it is necessary to plan an evaluation, monitoring and management of this data and also its risks taking into account its criticality.To the set of the above mentioned topics, we call it “data governance” and everyone in an organization is responsible, from the Board of Directors down, providing resources and decision-making, to the Analyst who feeds this data.
Data integrity is crucial in the implementation of a QMS and oncethis importance is ignored, it will not be possible to assure a robust system with effective, high-quality delivery of products or services.